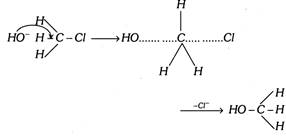
 In methyl chloride the - I effect of \[Cl\] group is further increased temporarily by the approach of hydroxyl ion.
In methyl chloride the - I effect of \[Cl\] group is further increased temporarily by the approach of hydroxyl ion. 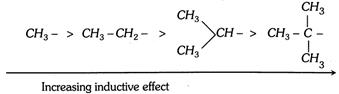 Electron donating power in decreasing order due to the hyperconjugation.
(6) Heat of hydrogenation : Hyperconjugation decreases the heat of hydrogenation.
(7) Dipole moment : Since hyperconjugation causes the development of charges, it also affects the dipole moment in the molecule.
The increase in dipole moment, when hydrogen of formaldehyde \[(\mu =2.27D)\] is replaced by methyl group, i.e., acetaldehyde \[(\mu =2.72D)\] can be referred to hyperconjugation, which leads to development of charges.
Electron donating power in decreasing order due to the hyperconjugation.
(6) Heat of hydrogenation : Hyperconjugation decreases the heat of hydrogenation.
(7) Dipole moment : Since hyperconjugation causes the development of charges, it also affects the dipole moment in the molecule.
The increase in dipole moment, when hydrogen of formaldehyde \[(\mu =2.27D)\] is replaced by methyl group, i.e., acetaldehyde \[(\mu =2.72D)\] can be referred to hyperconjugation, which leads to development of charges.
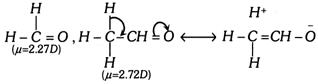 (8) Orienting influence of alkyl group in \[o,\,p\]-positions and of \[-CC{{l}_{3}}\] group in \[m\]-position : Ortho-para more...
(8) Orienting influence of alkyl group in \[o,\,p\]-positions and of \[-CC{{l}_{3}}\] group in \[m\]-position : Ortho-para more... 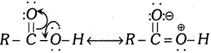 Both (i) and (ii) are the examples of mesomerism and resonance effect. Let us consider the following example.
Both (i) and (ii) are the examples of mesomerism and resonance effect. Let us consider the following example.
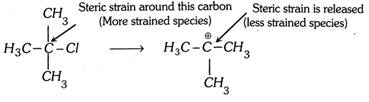 (2) Primary alkyl halide having quaternary \[\beta \]-carbon does not form transition state because of the steric strain around \[\alpha \]-carbon by the \[\beta \]-carbon. To release the strain it converts into carbocation.
(2) Primary alkyl halide having quaternary \[\beta \]-carbon does not form transition state because of the steric strain around \[\alpha \]-carbon by the \[\beta \]-carbon. To release the strain it converts into carbocation.
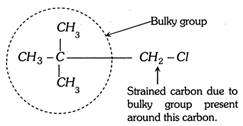 (3) Steric strain inhibits the resonance. This phenomenon is known as steric inhibitions of resonance.
(3) Steric strain inhibits the resonance. This phenomenon is known as steric inhibitions of resonance. | Type of hybridisation | \[s{{p}^{3}}\] | \[s{{p}^{2}}\] | \[sp\] | |||||||||||||||||||||||
| Number of orbitals used | \[1s\] and \[3p\] | \[1s\] and \[2p\] | \[1s\] and \[1p\] | |||||||||||||||||||||||
| Number of unused p-orbitals | Nil | One | Two | |||||||||||||||||||||||
| Bond | Four \[-\sigma \] | Three \[-\sigma \] One \[-\pi \] | Two \[-\sigma \] Two\[-\pi \] | |||||||||||||||||||||||
| more...
Werner was able to explain the bonding in complex.
Primary valency (Pv) : This is non- directional and ionizable. In fact it is the positive charge on the metal ion.
Secondary valency (Sv) : This is directional and non- ionizable. It is equal to the number of ligand atoms co-ordinated to the metal (co-ordination number). Example :
Compounds having the same molecular formula but different structures or spatial arrangements are called isomers and the phenomenon is referred as isomerism.
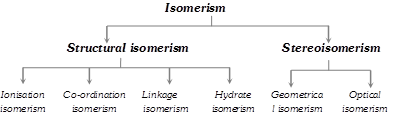 (1) Structural isomerism : Here the isomers have different arrangement of ligands around the central metal atom. It is of the following types :
(i) Ionisation isomerism : The co-ordination compound having the same composition or molecular formula but gives different ions in solution are called ionization isomers.
There is exchange of anions between the co-ordination sphere and ionization sphere.
(1) Structural isomerism : Here the isomers have different arrangement of ligands around the central metal atom. It is of the following types :
(i) Ionisation isomerism : The co-ordination compound having the same composition or molecular formula but gives different ions in solution are called ionization isomers.
There is exchange of anions between the co-ordination sphere and ionization sphere.
In order to name complex compounds certain rules have been framed by IUPAC. These are as follows :
(1) The positive part of a coordination compound is named first and is followed by the name of negative part.
(2) The ligands are named first followed by the central metal. The prefixes di-, tri-, tetra-, etc., are used to indicate the number of each kind of ligand present. The prefixes bis (two ligands), tris (three ligands), etc., are used when the ligands includes a number e.g., dipyridyl, bis (ethylenediamine).
(3) In polynuclear complexes, the bridging group is indicated in the formula of the complex by separating it from the rest of the complex by hyphens. In polynuclear complexes (a complex with two or more metal atoms), bridging ligand (which links two metal atoms) is denoted by the prefix \[\mu \] before its name.
(4) Naming of ligands : The different types of ligands i.e. neutral, negative or positive are named differently in a complex compound.
When a complex species has negative charge, the name of the central metal ends in - ate. For some elements, the ion name is based on the Latin name of the metal (for example, argentate for silver). Some such latin names used (with the suffix - ate) are given below :
|